Maintenance Notice (3:00 AM - 8:30 AM November 1, 2025 UTC): This website is scheduled to be unavailable due to maintenance. We appreciate your patience and understanding.
Product Document Searching Made Easy by 2D Code! | TCI Chemistry News October 2025 | [Product Highlights] Tetraphenylethylene Derivatives Used for... | Various analytical charts can be searched on each product detail page and Product Document Search (The kinds of analytical charts differ by product)
Maximum quantity allowed is 999
Please select the quantity
Chemistry Chat
Synthetic Uses of Molecular Iodine as an Oxidant
Hideo Togo
Emeritus Professor of Chiba University, Research Advisor in GODO SHIGEN Co. LTD
Molecular iodine (I2) is a mild oxidant for organic compounds. Therefore, treatment of thiols with molecular iodine in the presence of pyridine (Py) in chloroform at room temperature rapidly generates the corresponding disulfides quantitatively, as shown in Eq. 2.1. This reaction is an excellent and useful method for the preparation of disulfides from thiols. Generally, aromatic thiols are more rapidly oxidized to diaryl disulfides than aliphatic thiols due to the pKa difference between aromatic thiols and aliphatic thiols. For example, the pKa of thiophenol is 8.3, while that of butanethiol is 11.5. Solutions containing molecular iodine, such as iodine tincture and Isodine® (povidone iodine), have been used for sterilization of wounds and as mouthwashes. These medical effects are derived from the oxidation of cysteine in peptides to cystine by molecular iodine, changing the structures of the peptides.


Treatment of aldehydes with molecular iodine in the presence of KOH in MeOH at room temperature smoothly generates the corresponding methyl esters, as shown in Eq. 2.2.1) This reaction proceeds through formation of an O-I bond in the in situ generated hemiacetal, and then 1,2-elimination of HI to form the methyl ester.


Heat treatment of primary alcohols with molecular iodine in the presence of K2CO3 in MeOH generates the corresponding methyl carboxylates, as shown in Eq. 2.3.2) This reaction proceeds through 1,2-elimination of HI from in situ generated O-alkyl hypoiodite (RCH2O-I) to form an aldehyde, formation of a hemiacetal with MeOH, formation of an O-I bond in the hemiacetal, then the 1,2-elimination of HI to form the methyl carboxylate. Moreover, heat treatment of primary alcohols with molecular iodine in the presence of K2CO3 in tBuOH generates the corresponding oxidatively condensed carboxylate esters, as shown in Eq. 2.4.
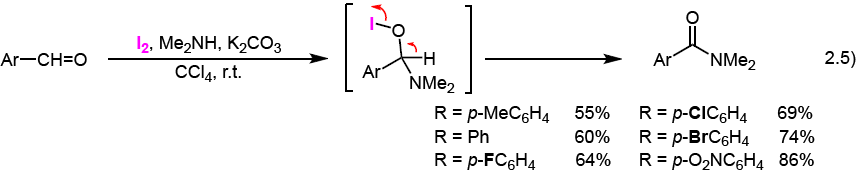
Treatment of aromatic aldehydes with Me2NH and molecular iodine in the presence of K2CO3 in CCl4 generates the corresponding N,N-dimethyl aromatic amides, as shown in Eq. 2.5.3) This reaction proceeds through the formation of an O-I bond in the in situ generated hemiaminal, and then 1,2-elimination of HI to form the aromatic amide. The same treatment of aliphatic aldehydes, instead of aromatic aldehydes, does not generate aliphatic amides due to the formation of enamines.
Heat treatment of primary alcohols with aq. NH3 (28% - 30%, same as below) and molecular iodine generates the corresponding nitriles, keeping the same number of carbon atoms, as shown in Eq. 2.6a.4) This reaction proceeds through 1,2-elimination of HI from in situ generated O-alkyl hypoiodite (RCH2O-I) to form an aldehyde, formation of an imine with NH3, formation of an N-I bond in the imine, and then 1,2-elimination of HI to form the nitrile. When the reactivities of molecular iodine, iodine chloride (ICl), N-bromosuccinimide (NBS), and N-chlorosuccinimide (NCS) with p-(methoxy)benzylalcohol and aq. NH3 (28% - 30%) are compared, reactions with molecular iodine, ICl, and NIS generate p-(methoxy)benzonitrile in good yields, as shown in Eq. 2.6b. However, reactions with NBS and NCS do not proceed at all. Thus, these results are derived from the specific character of the iodine atom.

Heat treatment of primary amines with aq. NH3 and molecular iodine generates the corresponding nitriles, keeping the same number of carbon atoms, as shown in Eq. 2.7a.4) This reaction proceeds through 1,2-elimination of HI from in situ generated N-iodoamine (RCH2NH-I) to form an imine, formation of N-(iodo)imine from the imine, and then 1,2-elimination of HI to form the nitrile. When the reactivities of molecular iodine, 1,3-diiodo-5,5-dimethylhydantoin (DIH), NIS, NBS, and NCS with dodecylamine and aq. NH3 are compared, reactions with molecular iodine, DIH, and NIS generate dodecanenitrile (lauronitrile) in good yields, as shown in Eq. 2.7b. However, reactions with NBS and NCS do not proceed at all.


The same treatment of aliphatic secondary amines and tertiary amines with aq. NH3 and excess amounts of molecular iodine generates 2 equivalents and 3 equivalents of the corresponding nitriles in good yields, respectively.
Heat treatment of benzylic halides with aq. NH3 and molecular iodine generates aromatic nitriles, as shown in Eq. 2.8a.5) The reactivity of benzylic halides decreases as follows: ArCH2I > ArCH2Br > ArCH2Cl. Here, the first reaction intermediate is benzylic amine. On the other hand, when aliphatic primary halides are used, a two-step process is required. Thus, heat treatment of aliphatic halides with aq. NH3, followed by reaction with molecular iodine and aq. NH3 under the same heat conditions generates the corresponding aliphatic nitriles, as shown in Eq. 2.8b.
Heat treatment of 1-(methoxy)naphthalene and 2-(methoxy)naphthalene, electron-rich aromatics, with POCl3 and DMF, followed by reaction with molecular iodine and aq. NH3 at room temperature, generates 4-cyano-1-(methoxy)naphthalene and 1-cyano-2-(methoxy)naphthalene in good yields, respectively, as shown in Eq. 2.9.6) Here, the first step proceeds through the Vilsmeier-Haack reaction, and the resulting N,N-(dimethyl)iminium salt reacts with NH3 and molecular iodine to form the aromatic nitrile. This reaction is a cyanide-free and metal-free method for the preparation of aromatic nitriles from aromatics. The same treatment of indole, thiophene, pyrrole, and furan with POCl3 and DMF, followed by reaction with molecular iodine and aq. NH3, generates 3-(cyano)indole, 2-(cyano)thiophene, 2-(cyano)pyrrole, and 2-(cyano)furan in good yields, respectively.

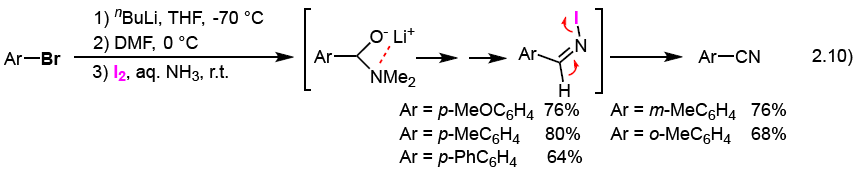
Treatment of aryl bromides (ArBr) with nBuLi in THF at low temperature, followed by reaction with DMF, and then with molecular iodine and aq. NH3 at room temperature, generates the corresponding aromatic nitriles, as shown in Eq. 2.10.7) Aryl iodides instead of aryl bromides, can also be used for the same reaction. By using this method, anisole, fluorobenzene, indole, benzothiophene, benzofuran, thiophene, and furan can be converted into the corresponding ortho-(cyano)aromatics or α-(cyano)heteroaromatics in good yields, respectively. Moreover, treatment of aryl bromides (ArBr) and alkyl bromides (RBr) with Mg in THF at room temperature, followed by reaction with DMF, and then with molecular iodine and aq. NH3 at room temperature, generates the corresponding aromatic nitriles and aliphatic nitriles, respectively, as shown in Eq. 2.11.8) Large-scale preparation of aromatic nitriles with a 200 L reactor by using this method in a chemical company could be successfully carried out.

Heat treatment of 2-cyclohexen-1-ones and cyclohexan-1,3-diones with molecular iodine in MeOH generates anisoles and 1,3-dimethoxybenzenes, respectively, as shown in Eq. 2.12a and Eq. 2.12b.9) These reactions proceed through the formation of hemiacetals with MeOH, dehydration of the hemiacetals by in situ generated HI, and then oxidation to the benzene ring by molecular iodine.


Irradiation treatment of a cyclic o-(diphenyl)benzene derivative in the presence of a catalytic amount of molecular iodine in benzene at room temperature under air with a Hg lamp generates the corresponding cyclic phenanthrene derivative in good yield, as shown in Eq. 2.13.10) The reaction proceeds through a conrotatory 6π-1,3,5-triene electrocyclic reaction under photolytic conditions to form a dibenzo-1,2-dihydrobenzene unit, which is then oxidized to a phenanthrene unit by molecular iodine. As mentioned above, molecular iodine has a moderate and unique oxidizing ability. These reactivities arise from the high polarizability and moderate electrophilicity of molecular iodine, and the high leaving ability and high nucleophilicity of the iodide anion.11) Therefore, the reactions described here are not applicable to molecular bromine (Br2), molecular chlorine (Cl2), NBS, or NCS.
* Isodine® is a trademark of iNova Pharmaceuticals Pte. Limited.
References
- 1) H. Abe, S. Itaya, K. Sasaki, T. Kobayashi, H. Ito, Chem. Commun. 2015, 51, 3586.

- 2) N. Mori, H. Togo, Tetrahedron 2005, 61, 5915.
- 3) H. Baba, K. Moriyama, H. Togo, Synlett 2012, 23, 1175.
- 4) S. Iida, H. Togo, Tetrahedron 2007, 63, 8274.
- 5) S. Iida, R. Ohmura, H. Togo, Tetrahedron 2009, 65, 6257.
- 6) S. Ushijima, K. Moriyama, H. Togo, Tetrahedron 2012, 68, 4588.
- 7) S. Ushijima, K. Moriyama, H. Togo, Tetrahedron 2011, 67, 958.
- 8) G. Ishii, R. Harigae, K. Moriyama, H. Togo, Tetrahedron 2013, 69, 1462.
- 9) Y. Jin, P. V. Petrovic, S. Huang, S. Banerjee, A. Nandy, P. T. Anastas, J. C. Lam, J. Org. Chem. 2024, 89, 3226.
- 10) T. Yamato, S. Miyamoto, T. Hironaka, Y. Miura, Org. Lett. 2005, 7, 3.
- 11) Hideo Togo, “Organic Iodine Chemistry” (Japanese), Kagaku-Dojin Publishing Co. (2005).
Introduction of the author
Hideo Togo
Hideo Togo was born in 1956 in Ibaraki prefecture of Japan. He completed his doctoral thesis in 1983 at University of Tsukuba. Then, he became a post-doctoral fellow at University of Lausanne in Switzerland (1983–1984) and at CNRS (Professor Sir Derek H. R. Barton) in France (1984–1985). Then, he became a research associate at University of Tsukuba in 1987 and then moved to Chiba University in 1989 as a research associate. He became an associate professor in 1994, and a full professor in 2005. He retired from Chiba University in 2021, and became an emeritus professor in Chiba University and a research advisor in research center of technology of Godo Shigen Co. LTD.